5. 分子動力学計算¶
分子動力学計算は以下の手順で行います。
Amberがインストールされているディレクトリはロードしたバージョンによって異なりますが、環境変数としてはすべて、$AMBERHOMEに設定されます。
このディレクトリ下の主なディレクトリを以下に示します(例ではamber/16up10)。
$AMBERHOME/内の主なディレクトリ
| ディレクトリ | 説明 |
|---|---|
| dat/ | Amberのデータベース |
| examples/ | デモンストレーションデータ |
| doc/ | マニュアルのファイル |
| bin/ | Amber実行モジュール |
| test/ | 各モジュールを実行するためのスクリプト例 |
Minimization、equilibration、Molecular Dynamicsは、binディレクトリ内にある sander というモジュールで行います。sander の使用方法は下のようになっています。
usage: sander [-O|A] -i mdin -o mdout -p prmtop -c inpcrd -r restrt
[-ref refc -x mdcrd -v mdvel -e mden -frc mdfrc -idip inpdip -rdip rstdip -mdip mddip
-inf mdinfo -radii radii -y inptraj -amd amd.log -scaledMD scaledMD.log] -cph-data <file>
それぞれのファイルは以下の通りです。
sanderによる計算に必要なファイル
| ファイル | 説明 |
|---|---|
| mdin | min/mdにおけるコントロールデータ(input)。 |
| mdout | アウトプットファイル(output)。 |
| prmtop | 分子のトポロジー、パラメータの情報のデータ(input)。 |
| inpcrd | 初期構造と(オプションで)初期速度のデータ(input)。 |
| restrt | 最終の座標、速度、ボックスサイズ(for Constant Pressure)のデータ(output)。 |
| refc | ポジションを束縛するときのデータ(input)。 |
| mdcrd | 座標値のトラジェクトリファイル(output)。 |
| mdvel | 速度のトラジェクトリファイル(output)。 |
| mden | エネルギーのトラジェクトリファイル(output)。 |
| mdinfo | mdoutの一番最新の部分(output)。 \ |
インプットファイルのオプション指定は&cntrlから/の間に記載し、オプションが複数ある場合は「,」で区切り、&cntrl から/までの行は必ず行頭に空白を入れて下さい。
インプットファイルで使用できる全オプションの詳細説明はマニュアルを参照して下さい。
インプットファイルのオプション
| オプション | 説明 |
|---|---|
| imin | 0の場合はMD計算、1の場合はエネルギー極小化計算、5の場合は振動解析を行います。 |
| nmropt | 0の場合はNMRを考慮しない計算、1,2の場合はNMR計算を行います。 |
| ntx | 初期座標、速度および基本セルの読み込み方法の指定フラグです。1がデフォルトとなっており、初期座標のみが呼び出されます。MD計算のリスタートでは5を利用して、初期座標、速度を呼び出します。基本セルはntb>0のときに行われます。なお、irestが1のときのみ、速度が呼び出されます。 |
| irest | リスタートフラグです。0の場合はリスタートを行わず、1の場合は座標や速度をリスタートファイルから読み込みます。 |
| ntpr | mdoutおよびmdinfoを更新するステップ数、デフォルトは50 |
| ntb | 周期的境界条件の設定 |
| cut | カットオフ値、単位はオングストローム |
| maxcyc | 極小化計算の最大サイクル数 |
| ntr | 0より大きい場合に位置束縛条件の設定を行います。デフォルトは0 |
| ntc | SHAKE法による分子固定設定、ntfと合わせて利用します。 |
| ntf | 力の評価設定、ntcと合わせて利用します。 |
| tempi | 初期温度設定 |
| temp0 | 定常温度の設定 |
| gamma_ln | 衝突頻度因子 |
| ntwx | mdcrd |
| ntwr | リスタートファイルを更新するステップ数、デフォルトはnstlim |
| nstlim | MD計算のステップ数 |
| pres0 | 初期圧力設定、デフォルトは1.0 |
| taup | 圧力緩和時間、デフォルトは1.0 |
| ntt | 定温シミュレーションの条件 |
| dt | トラジェクトリー計算における時間ステップ |
5.1. Minimization(構造最適化)¶
5.1.1. Minimizationのインプットファイル¶
#mdin
#minimization (minimizationの実行)
&cntrl
imin = 1, maxcyc = 200,ntpr = 10,
/
以下の実行項目を実施する場合はmdinという名称で上記サンプルを保存してください。
5.1.2. Minimizationの実行(インタラクティブ実行)¶
qrshコマンドで計算ノードにログイン後に以下のコマンドを実行してください。
*サンプルでは最小化サイクルの上限までまわるため、最小化計算は完了しません。
参考エネルギー極小化するためには14223回のステップが必要となります。
$ cp $AMBERSAMPLE/dna_pol/* ./
$ ls
inpcrd inpdip mdin prmtop
$ sander -i mdin -o mdout -p prmtop -c inpcrd
$ ls
inpcrd inpdip mdin mdinfo mdout prmtop restrt
並列計算を行う場合は下記コマンドを実行ください。
なお、サンプルは28並列計算時に5分以内で終了します。
$ mpirun -np [並列数] sander.MPI -O -i mdin -o mdoutp -p prmtop -c inpcrd
計算の詳細を確認する場合はmdoutファイル、概要を確認する場合はmdinfoを確認ください。
5.2. 溶媒を考慮したMM計算(構造最適化)¶
以下のサンプルはAmberのチュートリアルを元にTSUBAME3環境にあわせて各計算は10分以内に終了するように作成しております。
溶媒を考慮する場合は様々な手法がありますが、本書では、溶質を固定した計算(Step1)、全体計算(Step2)を行います。
計算時間は各手法ともに並列化なしで5分程度となります。その他手法については各自論文をご確認ください。
qrshコマンドで計算ノードにログイン後に以下のコマンドを実行してください。(以下のサンプルではAmber10Update10を利用します)
*ログインノードでの計算実行は行わないでください。
$ module load amber/16up10
サンプルファイルをダウンロードし、スクリプトを実行します。
$ cp $AMBERSAMPLE/dna_wat/step1/* ./
$ sh step1.sh
$ cp $AMBERSAMPLE/dna_wat/step2/* ./
$ sh step2.sh
step2.shを実行すると2つのPDBファイルが生成されます。
生成されたPDBを端末にダウンロードすることでBIOVIA DiscoveryStudio等の可視化ツールにて確認ができます。
下図はBIOVIA DiscoveryStudioで可視化した溶質固定後の計算結果、固定化を外した全体計算の結果、両者の重ね合わせを示します。
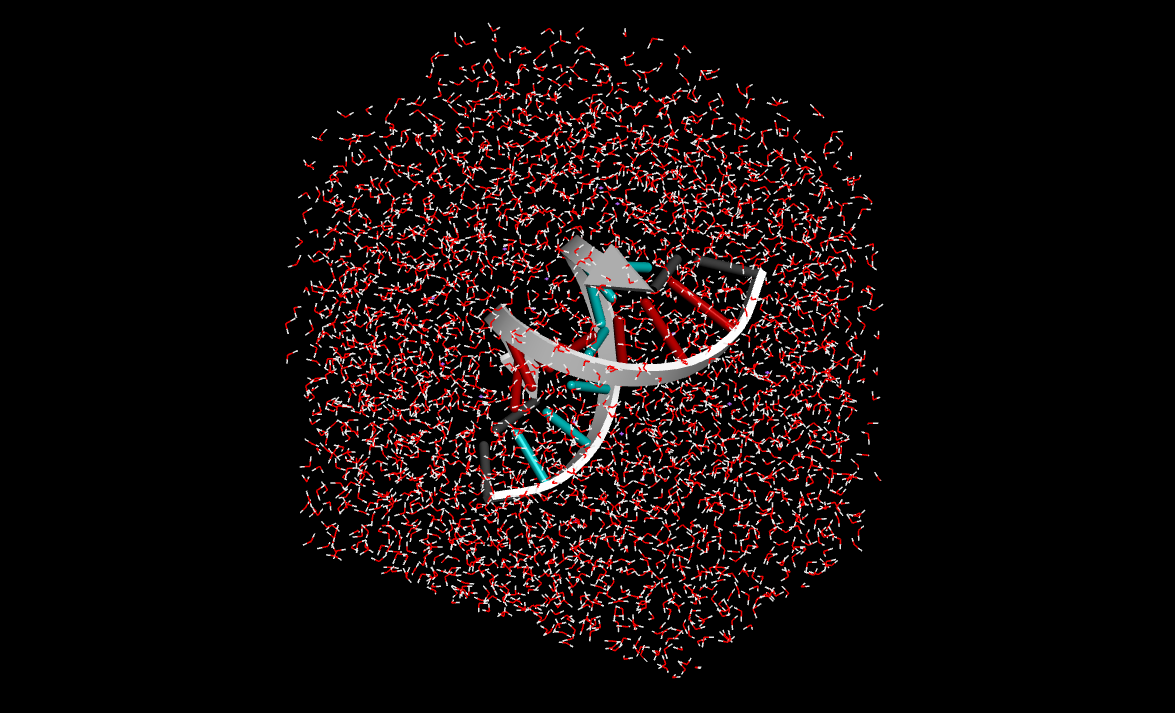
**溶質を固定した計算結果**
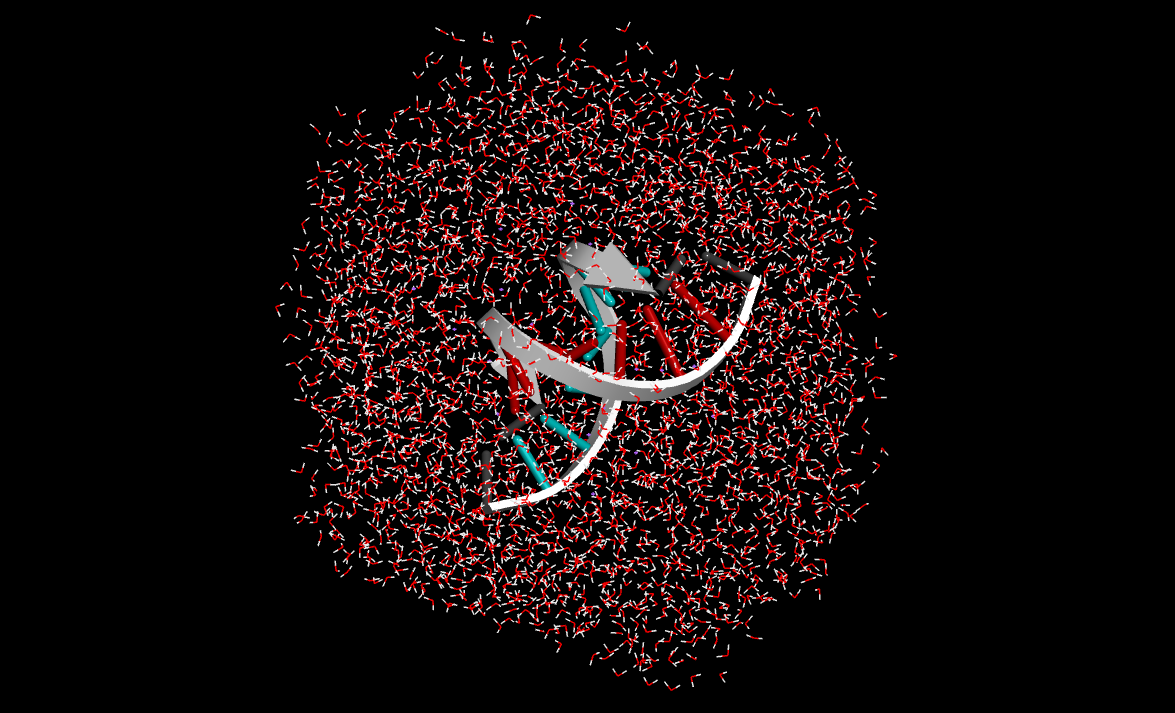
**固定化を外した全体計算の結果**
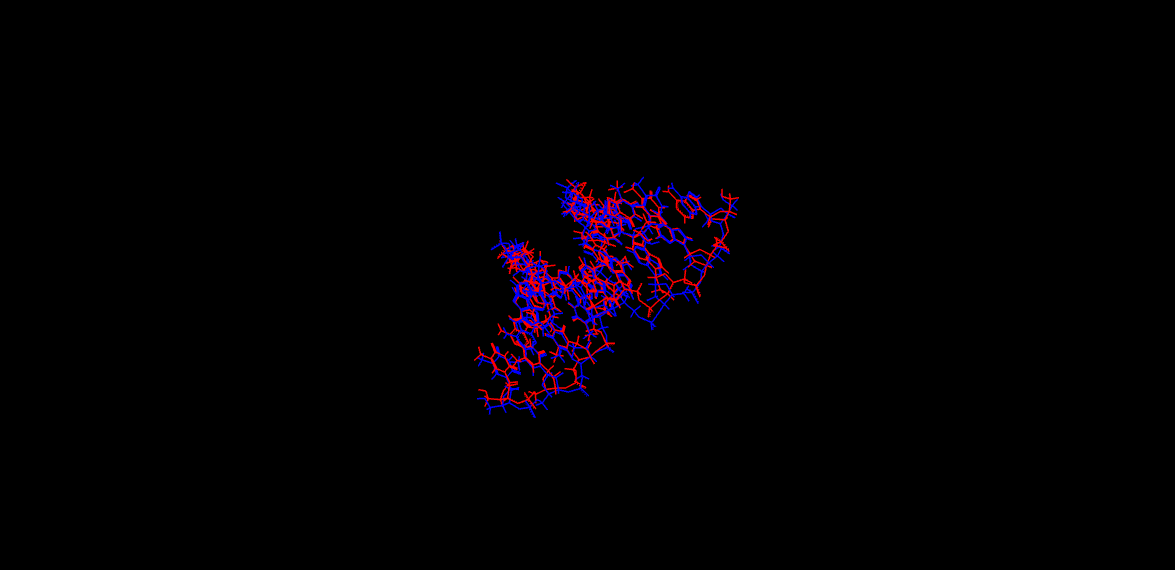
**溶質固定後の計算結果(赤)、固定化を外した全体計算(青)の結果の重ね合わせ(DNAのみ)**
5.3. 溶媒を考慮したMD計算(CPU/GPU)¶
5.3.1. ヒーティングおよび平衡化¶
MD計算時にも溶質の拘束計算後(Step3)に拘束を解いて全体計算(Step4)を行います。
また、Step3の20psでヒーティングを行い、Step4の100psで平衡化を行います。
Step3が並列化なしで6分、Step4は2ノード、CPU56並列でsander.MPIでは8分以上かかりますが、並列化効率の高いpmemd.MPIでは3分、cudaによる2ノード8GPU並列(pmemd.cuda.MPI)では2分以内に計算が終了します。
Step4ではqsubコマンドをご利用ください。
なお、下記のサンプル計算を行う場合はstep1,2の計算結果が必要となります。
pmemdは真空条件での計算はできませんので、ご注意ください。
非並列オプション実行時
$ cp $AMBERSAMPLE/dna_wat/step3/* ./
$ sh step3.sh
並列オプション実行時
$ cp $AMBERSAMPLE/dna_wat/step3/* ./
$ sh step3_parallel.sh
STEP4の並列計算(2ノード/CPU56並列利用)
$ cp $AMBERSAMPLE/dna_wat/step4/* ./
$ qsub step4_bacth.sh
STEP4の並列計算(CPU2ノード/8GPU並列利用)
$ cp $AMBERSAMPLE/dna_wat/step4/* ./
$ qsub step4_GPUbacth.sh
5.3.2. 結果ファイルの確認¶
Molecular Dynamics計算の出力ファイルmdoutファイルの内容を見て、計算結果を確認して下さい。 mdoutの主な内容は、出力された系の温度や圧力、エネルギー、体積などの情報です。 ここでは例としてCPUで計算した場合のstep4の計算結果の一部を表示します。
NSTEP = 40000 TIME(PS) = 80.000 TEMP(K) = 296.73 PRESS = 96.5
Etot = -28367.6754 EKtot = 5896.0101 EPtot = -34263.6855
BOND = 148.7758 ANGLE = 359.7502 DIHED = 458.8581
1-4 NB = 167.4830 1-4 EEL = -380.6307 VDWAALS = 3886.5010
EELEC = -38904.4229 EHBOND = 0.0000 RESTRAINT = 0.0000
EKCMT = 2718.6134 VIRIAL = 2514.8148 VOLUME = 97839.7312
Density = 1.0402
Ewald error estimate: 0.2648E-03
------------------------------------------------------------------------------
A V E R A G E S O V E R 40000 S T E P S
NSTEP = 40000 TIME(PS) = 80.000 TEMP(K) = 297.10 PRESS = -104.6
Etot = -28396.2451 EKtot = 5903.2761 EPtot = -34299.5212
BOND = 154.1835 ANGLE = 341.9533 DIHED = 458.8789
1-4 NB = 168.4548 1-4 EEL = -360.6335 VDWAALS = 3954.1168
EELEC = -39016.4751 EHBOND = 0.0000 RESTRAINT = 0.0000
EKCMT = 2722.3002 VIRIAL = 2965.6486 VOLUME = 98706.1131
Density = 1.0319
Ewald error estimate: 0.1054E-03
------------------------------------------------------------------------------
R M S F L U C T U A T I O N S
NSTEP = 40000 TIME(PS) = 80.000 TEMP(K) = 19.45 PRESS = 402.2
Etot = 724.1996 EKtot = 386.4596 EPtot = 352.5658
BOND = 11.1978 ANGLE = 15.7098 DIHED = 10.5140
1-4 NB = 4.9522 1-4 EEL = 13.0426 VDWAALS = 201.2018
EELEC = 526.0166 EHBOND = 0.0000 RESTRAINT = 0.0000
EKCMT = 176.9888 VIRIAL = 816.2426 VOLUME = 2935.8983
Density = 0.0276
Ewald error estimate: 0.8083E-04
主なパラメータの内容を以下に示します
mdoutのパラメータ
| パラメータ | 説明 |
|---|---|
| NSTEPS | ステップ数 |
| TIME(PS) | MDシミュレーション時間 |
| TEMP(K) | 温度 |
| PRESS | 圧力 |
| Etot | トータルエネルギー(単位はkcal/mol)( =EKtot + Eptot ) |
| EKto | 運動エネルギー |
| EPtot | ポテンシャルエネルギー( = BOND + ANGLE + DIHED + 1-4NB + 1-4EEL + VDWAALS + ELEC + EHBOND) |
| BOND | 結合の弾性エネルギー |
| ANGLE | 角度の弾性エネルギー |
| DIHED | 二面角によるエネルギー |
| 1-4 NB | 1-4非結合相互作用エネルギー |
| 1-4 EEL | 1-4静電エネルギー |
| VDWAALS | 非結合相互作用エネルギー |
| EELEC | 静電エネルギー |
| EHBOND | 水素結合のエネルギー |
| VOLUME | 体積(単位はA3) |
| Density | 密度(単位はg/cm3) |
5.3.3. mdout_analyzer.py¶
5章で計算を行ったサンプルデータを利用してmdout_analyzer.pyの利用方法をご説明します。
サンプルデータを読み込み、起動する場合は下記コマンドを実行ください。
$ mdout_analyzer.py dna_water_min.out
自分が計算したデータを解析したい場合は下記コマンドを実行ください。
$ mdout_analyzer.py <解析したいmdoutファイル>
コマンドを実行すると下記の画面が立ち上がります。
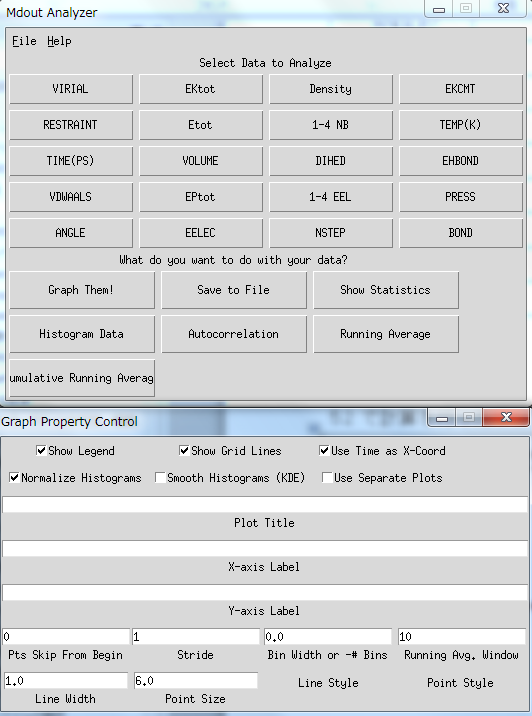
**mdout_analyzer起動画面**
グラフを作成するにはMdoutAnalyzerのSelect Data to Analyzeよりグラフ化したいボタンを選択し、GraphThem!をクリックしてください。 下図は5.1で計算した構造最適化におけるエネルギーの極小化グラフです。
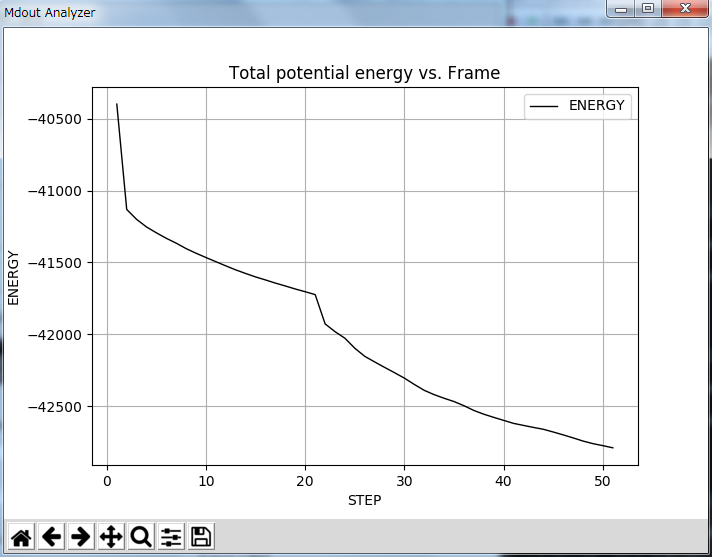
**Step毎のエネルギーの推移**